Nature | 人类基因组数据库帮助识别疾病变异

今天为大家介绍的是自Ryan S. Dhindsa & Slavé Petrovski团队的一篇报告。科研人员为一个名为gnomAD的人类基因组数据库进行了扩展,现在包含了76,156个完整的基因组序列。这个扩展版的数据库使得科学家能够研究基因组中非编码蛋白质区域的变异是如何影响人类健康的。

科学家们长期以来一直怀疑,许多引起疾病的遗传突变存在于不编码蛋白质的基因组的98%区域内,特别是在那些调控基因表达的区域。但要系统地区分有害突变和中性突变一直很有挑战性,部分原因是研究人员不清楚非编码基因组哪些部分对人类健康至关重要。在《自然》杂志上,Chen等人提出了一个工具,用于分析大量人类基因组,以识别在突变时最有可能导致疾病的非编码区域。这项工作代表了最新版本的基因组聚合数据库(gnomAD),这是一个公开的人类遗传变异目录。gnomAD的第一个版本于2020年发布,包含了125,748人的蛋白质编码DNA序列数据和15,708人的完整基因组。自那以后,该联盟大大扩展了这个数据库;现在的资源包括了76,156个不同血统个体的完整基因组序列,提供了更深入的人类遗传变异图景。
gnomAD已经改变了人类遗传学领域,尤其是在诊断罕见疾病方面。任何个体的基因组与其他人相比都有数百万处不同。这些遗传变异中的大多数在临床上是无关紧要的,特别是那些在普通人群中很常见的变异。当临床遗传学家分析一个疑似患有罕见遗传病的人的DNA时,他们必须筛选所有变异,排除那些常见的,从而找到最有可能导致疾病的变异。gnomAD使临床医生能够轻松地查找一个人的变异,并排除那些在全球范围内或在某种遗传血统中很常见的变异。这个最新版本的gnomAD中更大且血统更多样化的完整基因组将使科学家更容易识别哪些变异在非编码基因组中是罕见的——因此更有可能与疾病相关。
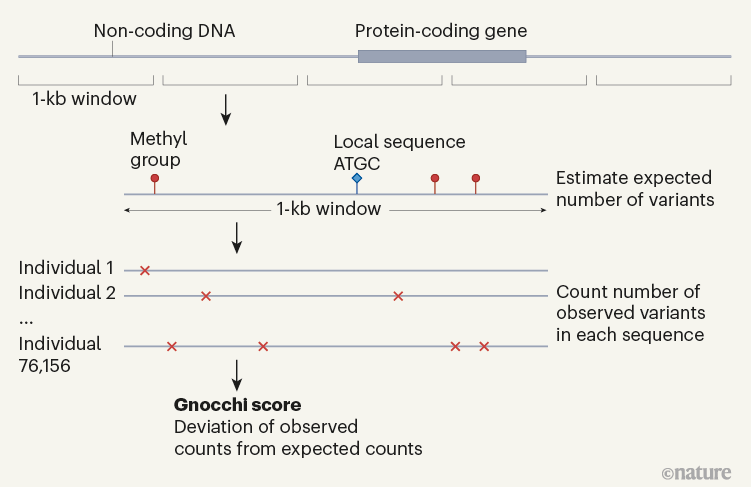
图 1
gnomAD这样的大型数据集使研究人员能够开发出所谓的“不耐受度量指标”,来检查在人类大样本中观察到多少改变蛋白质的基因变异,与在进化过程中随机出现的变异数量相比。这些措施有助于确定哪些基因不耐受遗传变异。与预期相比,变异较少的基因更不耐受——也更可能与疾病相关。为了包含人类基因组的非编码部分,越来越多的努力被投入到扩展不耐受度量指标上。Chen等人使用他们称为Gnocchi的方法,为这些工具增添了新的一项功能(图1)。与有明确边界的蛋白质编码基因不同,非编码区域并没有方便地划分为功能单元。为了解决这个问题,作者将基因组分为1000碱基对的窗口,并计算了每个窗口的不耐受度。
虽然Gnocchi在概念上与其他非编码不耐受度量指标相似,但它在计算每个窗口理论上预期的变异数量方面取得了重大进步。基因组中的突变率受到多种因素的影响,比如局部序列环境和DNA通过甲基化等方式的修改。Chen和同事引入了一个统计模型,包含了这些不同特征,以更好地估计每个窗口的突变率,并通过多种方式验证了Gnocchi识别基因组相关区域的能力。首先,他们展示了蛋白质编码区域平均比非编码区域更不耐受变异,这与预期一致。其次,他们发现非编码基因组中最不耐受的区域富含基因调控元素,如启动子和增强子。第三,他们证明Gnocchi能区分可能是良性的变异和在非编码基因组中策划的疾病引起的突变列表。第四,他们表明被诊断为发育障碍的个体比健康人更有可能在不耐受区域的基因组中有拷贝数变异(导致DNA重复或删除的大变异)。
Chen和同事还展示了Gnocchi可以用来加强传统的基因水平不耐受度量指标。他们将一个基因的非编码增强子对变异的不耐受(使用Gnocchi测量)与基因编码区域对破坏其正常功能的变异的不耐受(使用另一个叫做LOEUF的指标测量)进行了比较。这两个指标通常一致,但在某些情况下,比如一个看似耐受功能丧失的基因有一个不耐受的增强子。这些情况主要出现在小基因上,目前其基因水平不耐受度量指标的准确性受到样本大小的限制。将一个基因的LOEUF分数与其增强子的Gnocchi分数结合起来,提高了小蛋白质编码基因的整体不耐受估计。
研究人员展示了在将基因水平的不耐受度量指标与测量基因未翻译区域的不耐受度量指标相结合时的类似性能提升。通过结合每个基因及其附近和远处调控序列的不耐受度量指标,这种方法在未来的工作中可以进一步完善。值得注意的是,Gnocchi在识别非编码、与疾病相关的变异方面似乎比现有指标更有优势。Gnocchi更好的表现可能可以由其分数制定的差异和其对突变产生方式的建模来解释。然而,gnomAD收集的基因组序列包含的非欧洲血统个体比UK Biobank多,这也可能解释一些性能差异。值得称赞的是,gnomAD近一半的基因组样本来自非欧洲血统的个体,但研究人员必须继续努力获得更大、更多样化的人类参考集,以增加不耐受度量指标的准确性和改善健康公平。
gnomAD联盟在其第一次迭代中设定了数据聚合和共享的黄金标准,并在这方面继续树立典范。在gnomAD联盟的强大领导下,这一资源将继续增长,它已明确表示优先事项是不断扩大数据库,使其更具全球人口的代表性。这样做,它将为科学家提供更多工具来揭示我们基因组的隐藏秘密。
编译 | 曾全晨
审稿 | 王建民
参考资料
Ryan S. Dhindsa & Slavé Petrovski. An expanded genomic database for identifying disease-related variants. Nature.
doi: https://doi.org/10.1038/d41586-023-01598-6