细胞通讯分析之CellphoneDB(二)可视化篇

时间过得好快,又一个月过去了!前几期我分享了CellChat的学习笔记,包括:
在此基础上,我还写了CellphoneDB的笔记:细胞通讯分析之CellphoneDB初探(一),在这个帖子里简单介绍了CellphoneDB,以及CellphoneDB的环境配制、单样本实战,最后提供了一个可视化的函数cellphoneDB_Dotplot。另外,cellphoneDB似乎是不支持小鼠等其他物种的数据,因此我写了 一行代码完成单细胞数据人鼠基因同源转换,提供了一个函数,一行代码完成人鼠的基因同源转换,然后用转换后的数据走cellphoneDB流程即可。
本期的帖子接上文,主要解决CellphoneDB的2个问题:
- 由于目前一个单细胞项目基本包含数个/数十个样本,如何实现CellphoneDB的批量处理;
- 实现更多更丰富的可视化。
一. CellphoneDB的批量处理
1.1 首先在R语言环境下输出表达数据及表型数据:
library(Seurat)
library(tidyverse)
library(data.table)
library(dplyr)
library(readr)
#### 1.load data
scRNA = read_rds("./Step4.m.seurat.rds")
#### 2.批量输出Output
out.dir = "Step8.CellphoneDB/"
for(sample in unique(scRNA$sampleID)){
sp1 <- subset(scRNA,sampleID == sample)
sp1_counts <- as.data.frame(sp1@assays$RNA@data) %>%
rownames_to_column(var = "Gene")
sp1_meta <- data.frame(Cell=rownames(sp1@meta.data),
cell_type=sp1@meta.data$celltype)
fwrite(sp1_counts,file = paste0(out.dir,sample,"_counts.txt"), row.names=F, sep='\t')
fwrite(sp1_meta, file = paste0(out.dir,sample,"_meta.txt"), row.names=F, sep='\t')
}
运行之后out.dir目录下就会生成每个样本的表达数据和meta表型数据的txt文件。
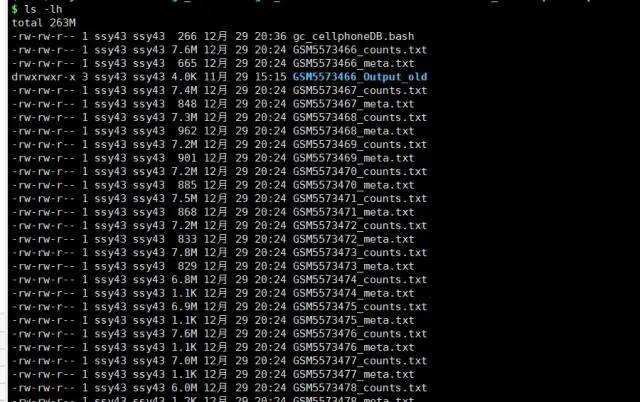
然后在Linux环境下批量运行CellphoneDB:
1.2 首先写一个通用型的shell脚本
##进入目的文件夹
cd Step8.CellphoneDB/
##先写个脚本
cat >gc_cellphoneDB.bash
file_count=./$1_counts.txt
file_anno=./$1_meta.txt
outdir=./$1_Output
if [ ! -d ${outdir} ]; then
mkdir ${outdir}
fi
cellphonedb method statistical_analysis ${file_anno} ${file_count} --counts-data hgnc_symbol --output-path ${outdir} --threshold 0.01 --threads 10
##如果细胞数太多,可以添加下采样参数,默认只分析1/3的细胞
#--subsampling
#--subsampling-log true #对于没有log转化的数据,还要加这个参数
1.3 然后批量运行
##激活环境
conda activate cpdb
##批量运行此脚本
ls *counts.txt | while read id;
do
id=${id%%_counts.txt}
echo $id
bash gc_cellphoneDB.bash $id 1>${id}.log 2>&1
done
每个样本的结果在当前目录下。
二. CellphoneDB的多种可视化方案
除了我上期用健明老师的代码改编的函数cellphoneDB_Dotplot绘制气泡图(详见细胞通讯分析之CellphoneDB初探(一)),我们还可以使用如下方案进行可视化。
2.1 衔接Cellchat的函数
其实只要深刻理解Cellchat这个包的函数逻辑,我们即可用cellphoneDB的结果作为input数据,进行可视化。
这里需要的文件是count_network.txt作为input数据。
library(CellChat)
library(tidyr)
df.net <- read.table("./Step8.CellphoneDB/GSM5573466_Output/Outplot/count_network.txt",
header = T,sep = "\t",stringsAsFactors = F)
meta.data <- read.table("./Step8.CellphoneDB/GSM5573466_meta.txt",
header = T,sep = "\t",stringsAsFactors = F)
groupSize <- as.numeric(table(meta.data$cell_type))
df.net <- spread(df.net, TARGET, count)
rownames(df.net) <- df.net$SOURCE
df.net <- df.net[, -1]
df.net <- as.matrix(df.net)
netVisual_circle(df.net, vertex.weight = groupSize,
weight.scale = T, label.edge= F,
title.name = "Number of interactions")

image-20221229204130690
# par(mfrow = c(3,3), xpd=TRUE)
for (i in 1:nrow(df.net)) {
mat2 <- matrix(0, nrow = nrow(df.net), ncol = ncol(df.net), dimnames = dimnames(df.net))
mat2[i, ] <- df.net[i, ]
netVisual_circle(mat2,
vertex.weight = groupSize,
weight.scale = T,
edge.weight.max = max(df.net),
title.name = rownames(df.net)[i],
arrow.size=0.5)
}
批量输出互作结果,这里只显示其中1个:
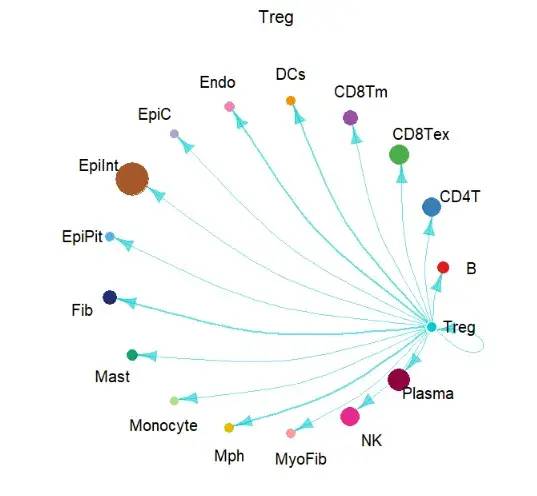
2.2 R包ktplots
参考追风哥在简书上写的笔记:10X单细胞 && 10X空间转录组分析之cellphoneDB可视化进阶,github: https://github.com/zktuong/ktplots。这个包自带了非常多的可视化方案,这里列举其中一两种:
#devtools::install_github('zktuong/ktplots', dependencies = TRUE)
library(ktplots)
library(Seurat)
library(SingleCellExperiment)
library(tidyverse)
library(data.table)
library(dplyr)
library(readr)
#### 1 加载数据
mypvals <- read.table("./Step8.CellphoneDB/GSM5573466_Output/pvalues.txt",header = T,sep = "\t",stringsAsFactors = F,check.names = F)
mymeans <- read.table("./Step8.CellphoneDB/GSM5573466_Output/means.txt",header = T,sep = "\t",stringsAsFactors = F,check.names = F)
mydecon <- read.table("./Step8.CellphoneDB/GSM5573466_Output/deconvoluted.txt",header = T,sep = "\t",stringsAsFactors = F,check.names = F)
#单细胞数据
seurat.data = read_rds("./Step4.m.seurat.rds")
scRNA = subset(seurat.data,sampleID == "GSM5573466")
table(scRNA$celltype)
#Seurat转SCE
sce <- Seurat::as.SingleCellExperiment(scRNA)
## Vis 1
p1 <- plot_cpdb3(cell_type1 = 'CD4T|Treg',
cell_type2 = 'EpiC|EpiInt|EpiPit',
scdata = sce,
idents = 'celltype', # column name where the cell ids are located in the metadata
means = mymeans,
pvals = mypvals,
deconvoluted = mydecon, # new options from here on specific to plot_cpdb3
keep_significant_only = TRUE,
standard_scale = TRUE,
remove_self = TRUE,
legend.pos.x = -5,legend.pos.y = 5
)
p1
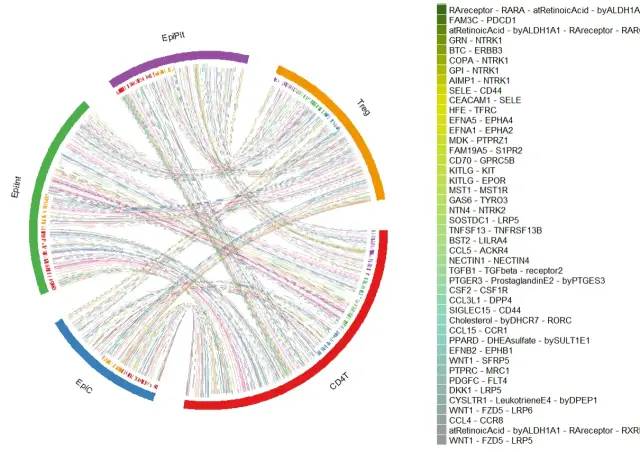
再复杂一点的:
p2 <- plot_cpdb2(cell_type1 = "CD4T|Treg", # same usage style as plot_cpdb
cell_type2 = 'EpiC|EpiInt|EpiPit',
idents = 'celltype',
scdata = sce,
means = mymeans,
pvals = mypvals,
deconvoluted = mydecon, # new options from here on specific to plot_cpdb2
desiredInteractions = list(c('CD4T', 'Treg'), c('Treg', 'EpiC'), c('Treg', 'EpiInt'), c('Treg', 'EpiPit')),
interaction_grouping = interaction_annotation,
edge_group_colors = c("Activating" = "#e15759", "Chemotaxis" = "#59a14f", "Inhibitory" = "#4e79a7", "Intracellular trafficking" = "#9c755f", "DC_development" = "#B07aa1"),
node_group_colors = c("CD4T" = "#86bc86", "Treg" = "#79706e", "EpiC" = "#ff7f0e", "EpiInt" = "#bcbd22" ,"EpiPit" = "#17becf"),
keep_significant_only = TRUE,
standard_scale = TRUE,
remove_self = TRUE)
p2
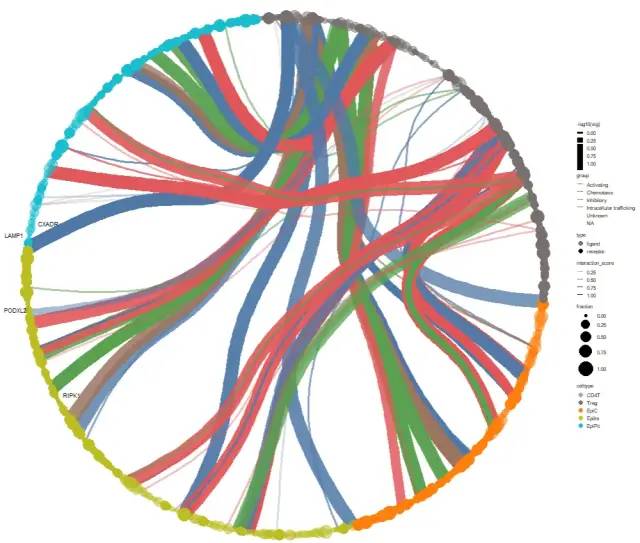
R包ktplots提供了非常多的可视化方案,详见github: https://github.com/zktuong/ktplot
- END -